HW3.Genome Assembly. Part 1
Ціль
Мапування визначених ліній E.coli на референтний геном з NCBI. Оцінка якості відсеквенованих зразків та оцінка глибини покриття
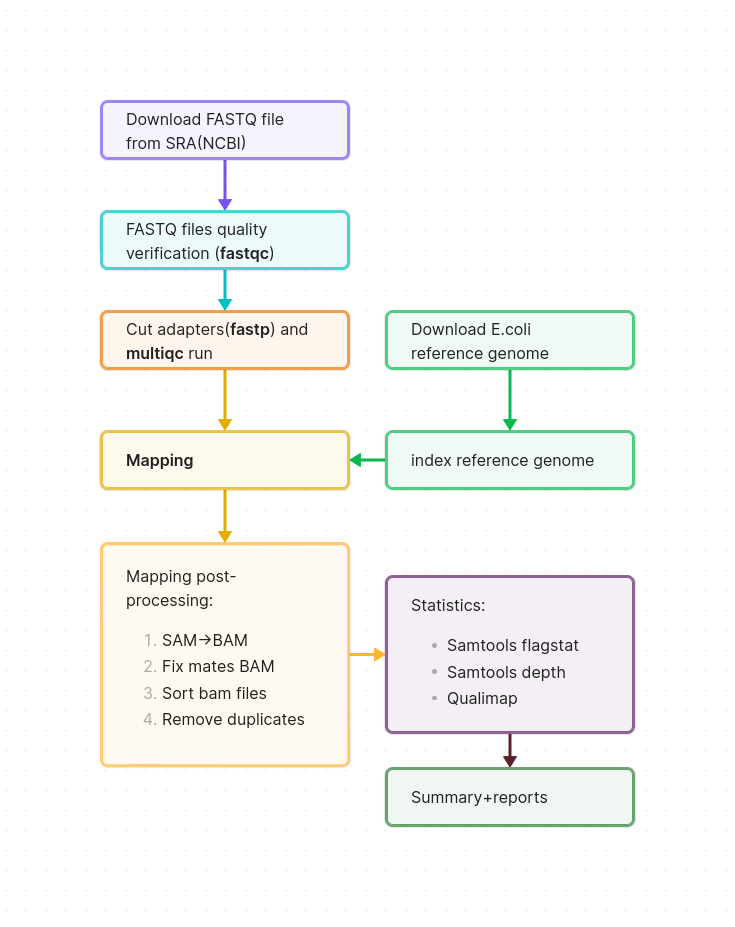
Пайплайн
SRA Accession List (одна лінія для кожної групи)
DRX569192(DRR588645)
DRX569193(DRR588646)
DRX569194(DRR588647)
DRX569195(DRR588648)
DRX569197(DRR588650)
DRX569196(DRR588649)
DRX569198(DRR588651)
З повним переліком SRA Accession List можна ознайомитись тут.
Крок 1. Завантаження рідів з бази даних SRA
# Створення папки для аналізу
mkdir Ecoli_analysis
# Відкрити папку з аналізом
cd Ecoli_analysis
#Створення середовища SRA
conda create -n SRA
# Активація середовища SRA
conda activate SRA
# Встановлення пакунку SRA-tools
conda install sra-tools
# створення папки data для зберігання даних секвенування
mkdir data | cd data
# Команда для завантаження FASTQ файлів у цільову папку data за допомогою інструменту fasterq-dump
fasterq-dump DRR589139Крок 2. Обрізка адаптерів за допомогою fastp
# Створення нового середовища qc і встановлення інструментів fastp fastqc multiqc:
conda create --yes -n qc fastp fastqc multiqc
# активувати середовище qc:
conda activate qc
# Перейти у попередню папку
cd ..
#створити папку trimmed:
mkdir trimmed
#Обрізка адаптерів за допомогою fastp:
fastp --detect_adapter_for_pe --overrepresentation_analysis --correction --cut_right --thread 2 --html trimmed/DRR589139.fastp.html --json trimmed/DRR589139.fastp.json -i data/DRR589139_1.fastq -I data/DRR589139_2.fastq -o trimmed/DRR589139_1.fastq -O trimmed/DRR589139_2.fastqКрок 3. Перевірка якості обрізаних рідів
# поверністься у папку Ecoli_analysis та створіть папку trimmed-fastqc
mkdir trimmed-fastqc
#Генерування репорту за допомогою інструменту FASTQC вашої лінії E.coli
fastqc -o trimmed-fastqc data/DRR589139_1.fastq data/DRR589139_2.fastq
#Run MultiQC
multiqc trimmed trimmed-fastqcКрок 4. Мапування на референтний геном
# Створення папки для мапування
mkdir mapping
cd mapping
# Завантаження референтного геному з ftp серверу NCBI
mkdir reference
cd reference
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/005/845/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_genomic.fna.gz
# Створити нове середовище mapping та інсталювати у ньому програми samtools, bwa, qualimap, r-base
conda create --yes -n mapping samtools bwa qualimap r-base
# Активувати нове середовище
conda activate mapping
# Розархівування даних в папці
gunzip GCF_000005845.2_ASM584v2_genomic.fna.gz
# Індексування
bwa index GCF_000005845.2_ASM584v2_genomic.fna
# Paired-end мапування проводиться в папці експерименту. У нашому випадку це папка data. Щоб перевірити шлях скористайтесь командою pwd щоб бути впевненим у правильному розташуванні перед запуском програми.
pwd
bwa mem mapping/reference/GCF_000005845.2_ASM584v2_genomic.fna trimmed/DRR589139_1.fastq trimmed/DRR589139_2.fastq > mapping/aln-DRR589139.samПост-процессинг мапування
# фіксування пар (mates) і стиснення файлів sam
samtools sort -n -O sam aln-DRR589139.sam | samtools fixmate -m -O bam - DRR589139.fixmate.bam
# видалення sam файлу для економії простору
rm aln-DRR589139.sam
# Сортування bam файлу по координатам
samtools sort -O bam -o DRR589139.sorted.bam DRR589139.fixmate.bam
# Видалення дуплікацій
samtools markdup -r -S DRR589139.sorted.bam DRR589139.sorted.dedup.bam
# Видалення попереднього fixmate і sorted файлу
rm DRR589139.fixmate.bam | rm DRR589139.sorted.bam
# Статистика відфільтрованого bam фалу
samtools flagstat DRR589139.sorted.dedup.bam
# Перевірка якості - оцінка глибини покриття зчитувань
samtools depth DRR589139.sorted.dedup.bam | gzip > DRR589139.depth.txt.gz
# Запуск qualimap для DRR589139.sorted.dedup.bam файлу
qualimap bamqc -bam DRR589139.sorted.dedup.bam